
Can AI help us trace Omicron’s origins?
(Probably not in this case)
Where did the new SARS-CoV-2 variant of concern, termed B.1.1.529 or Omicron, come from? Significant interest in Omicron stems from concerns about the possibilities of immune escape and higher transmissibility, but little is known about its origins so far. The virus is unusually divergent from other circulating lineages, and most closely related to sequences from mid-2020, raising questions about possible origins in either (a) a chronically-infected immunocompromised person or (b) a possible animal reservoir. SARS-CoV-2 has undergone “spillback” into farmed animals (mink), captive wildlife (lions, tigers, gorillas, and many other species), pets (cats and dogs), and wild animals (white-tailed deer), and experimental infections and modeling studies suggest even more species have the capacity to become infected. Based on mutations seen in different animals, some scientists have been speculating that rodents might have been the source of secondary spillover.
Could some of the tools that our team uses - specifically, ones that predict viral origins based on their genome composition biases - help solve this mystery?
Viruses co-evolve with different hosts in ways that sometimes leave behind recognizable signatures. Vertebrates, for example, have lower rates of CG dinucleotides in their own genomes, which their immune systems use as a kind of “calling card” to identify genetic material that isn’t their own; viral genomes, in turn, seem to use CG dinucleotides more sparingly in vertebrate hosts, helping them fly under the radar.
Evolution takes time, though, and occurs on different scales. Just as it’s easier to swap out one word of a paragraph than completely change its tone, point mutations happen faster than changes in genome composition bias. There may be some overlap between mutations in Omicron and those seen previously in SARS-CoV-2 in rodents, some of which might be adaptive (host-specific adaptation or otherwise), but over the timescale on which Omicron emerged, we probably wouldn’t expect genome composition bias to tell us much.
To illustrate the problem, we’ve applied the random forest machine learning models from Brierley & Fowler 2021, which are trained to predict host origin of coronaviruses from genome composition features (such as codon usage bias). In that study, we showed that models suggest a bat origin to SARS-CoV-2 with medium high confidence (specifically in the Yinpterochiroptera, a suborder that includes horseshoe bats, the reservoirs of most known SARS-like viruses). For comparison, the model was able to link MERS-CoV to camels and SARS-CoV to carnivores (i.e., civets; and secondarily, the Yinchiroptera) with high confidence, given an abundance of training data for each:
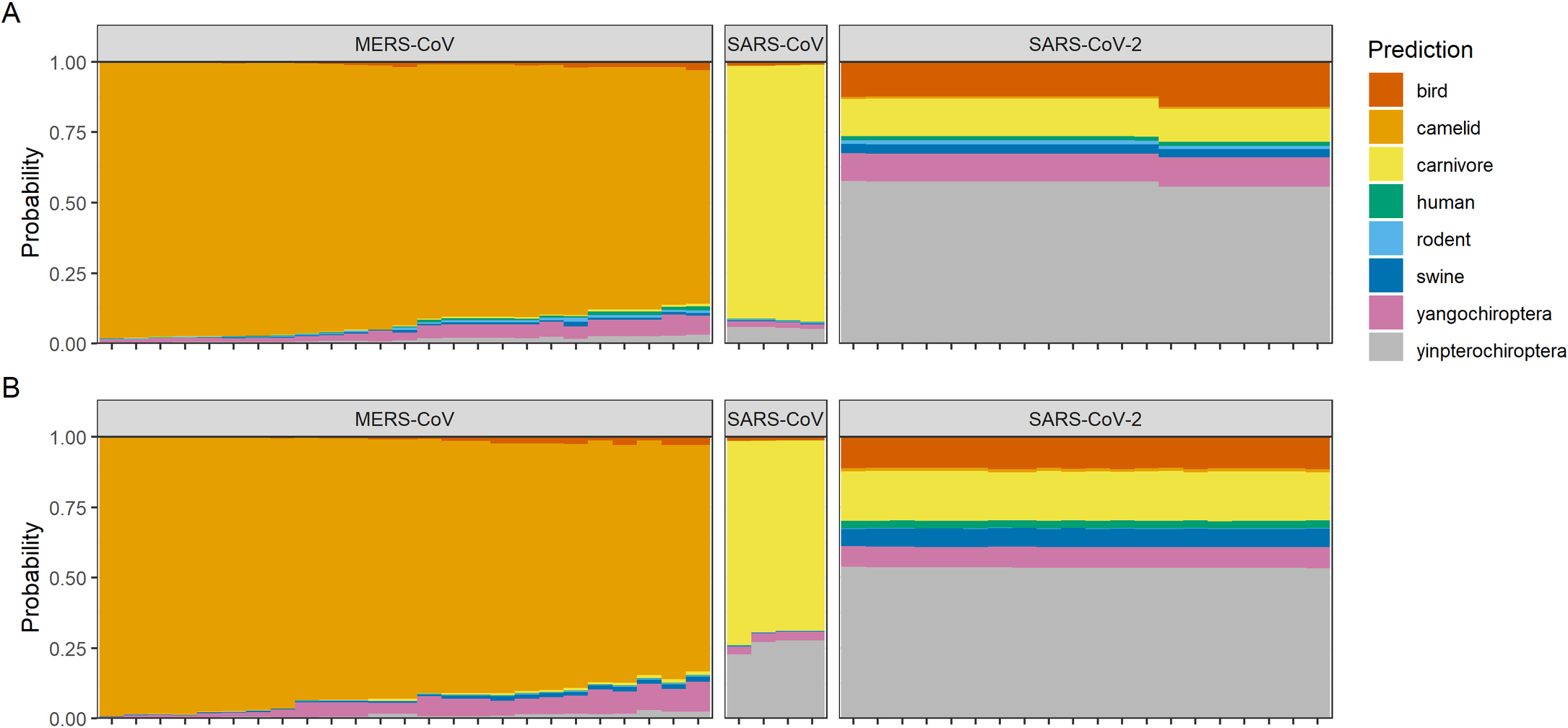
Reproduced from Brierley, Liam, and Anna Fowler. "Predicting the animal hosts of coronaviruses from compositional biases of spike protein and whole genome sequences through machine learning." PLoS Pathogens 17.4 (2021): e1009149.
Here, we’ve updated those predictions using whole genome sequences of each SARS-CoV-2 variant of concern. For reference, we’ve shown three additional viruses that show different levels of adaptation to rodent hosts:
A lab mouse-adapted SARS-CoV lineage (MA-15; somewhat host-adapted, but under evolutionary pressures that might or might not be different from those in wild hosts)
A recently discovered recombinant betacoronavirus found in Rattus rattus in China (rat coronavirus GCCDC4; unknown level of host adaptation, and notably not in the training data)
Three sequences of murine coronavirus (M-CoV; definitely host-adapted)
All sequences are publicly available on GenBank. Here’s the predictions:

And here’s the same results based on just spike gene sequences:

What does this show?
Since SARS-CoV-2 first emerged in humans, there’s been fairly minimal change in the signal of host adaptation recoverable from genome composition biases - including in Omicron, which isn’t recognizably more “rodent-shaped” than any other lineage
The mouse-adapted serially passaged SARS-CoV (MA-15) looks fairly similar to the other SARS-CoV predictions (see above), and isn’t successfully recognized by the model as rodent-adapted
On the other hand, the two natural rodent coronaviruses are successfully recognized by the model as such (independent of whether or not the model has seen sequences of these viruses before in the training data)
So: no evidence from these specific analyses that Omicron is any more “mousy” than any other lineage. But, this tool isn’t really fit for this specific purpose. As you can see in the difference between the other viruses that have adapted to rodent hosts in the lab versus in nature, timescales matter.
Genome compositional features can be informative about host-virus relationships that have developed over thousands of years of evolution. But by comparison, they tend to change very little over the course of any individual epidemic in novel host(s). In other words, we don’t (and wouldn’t expect to) see adaptation to humans in SARS-CoV-2’s dinucleotide and codon usage “in real time”, even if the virus had undergone spillback into rodents and secondary spillover back into humans.
That doesn’t mean individual mutations aren’t important for the virus’ phenotype and its ultimate epidemiology. (What we show here doesn’t change the tentative link between Omicron mutations and possible rodent hosts!) But what it does mean is that such models trained on signals at the macroevolutionary level are going to be uninformative (or at worst, misleading) to answer questions about how the pandemic is changing. Researchers should be careful to not oversell results on this front as they dig into Omicron’s origins. There’s only one way to get the definitive answer about where Omicron came from: rapid and coordinated efforts to collect human and wild animal sequences and fill in the “missing links.”
Meanwhile, there’s still plenty of mysteries to solve about pathogen spillback - for more on that, check out our evolving preprint on the subject.